Research themes
What is the interactome?
Although the human genome sequence was successfully determined in the genome project, the functions for more than half of the discovered genes (proteins) were unknown. To determine the protein functions, it is important to find their interaction partners. Proteins always interact with other biomolecules for their biological functions. We call the entirety of these interactions between biomolecules as the 'interactome'. However, to correctly understand dynamically changing interactome networks is not easy. New methods have had to be developed to analyze spatiotemporal networks efficiently. Our challenge is to develop these new technologies for capturing the spatiotemporal networks.
In the era of the personal genome, the main goal of our laboratory is to realize "interactome medical science" treatment, especially for cancer, using technology developed in Japan. We have three research themes to achieve this goal:
- Pioneering Technology – Pioneering Technology – Understanding the characteristics and associations of cancer cells using dynamic interactome network analysis.
- Mastering Technology – Developing innovative tools for meta-interactome analysis.
- Uzing Technology – Protein-domain targeted drug discovery using interacting regions (IR) data and the development of model mice for validation.
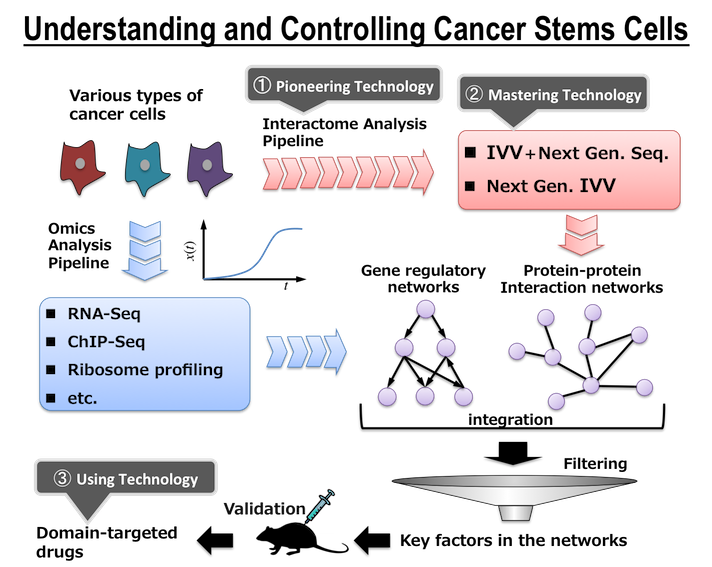
1. Pioneering Technology – Understanding the characteristics and associations of cancer cells using dynamic interactome network analysis.
We are attempting to understand cancer cells (especially cancer stem cells) as a system and to control them in collaboration with cancer researchers, interactome analysis researchers, and bioinformaticians who can analyze the massive amount of -omics data using supercomputers. To realize quantitative and accurate analysis of dynamically changing interaction networks, we aim to develop an interactome analysis pipeline using "puromycin technology", which was developed in Japan, as part of the analysis pipeline for cancer -omics. In addition, we are developing two databases, IRView (for protein interacting regions) and PRD (for protein-RNA interactions), as analysis platforms for the large amount of interaction data produced by next generation sequencing.
2. Mastering Technology – Developing innovative tools for meta-interactome analysis.
To correctly understand the individualities of cancer cells, their diversity must be determined by comparing various types of cancer cells. The next generation sequencing technology has more than 100,000 times higher throughput than the conventional method. Using this technology, we can simultaneously analyze various types of -omics data (e.g., genome, epigenome and transcriptome) for various types of cancer cells. However, it is not easy to realize the high-throughput interactome analysis by means of the next generation sequencer as a DNA analyzing device. Therefore, we are developing an innovative interactome analysis method using next generation sequencing for realizing the meta-interactome analysis (spatiotemporal interactome analysis) and for personal interactome analysis.
3. Using Technology – Protein-domain targeted drug discovery using interacting regions (IR) data and the development of model mice for validation.
To solve the side-effect problem in anticancer drugs, molecular-targeted drugs with smaller side effects have been developed. The protein-protein interaction data produced by our laboratory contain not only the data for interacting proteins but also data for interacting regions (IRs) of those proteins. These data are a potentially valuable resource for domain-targeted drugs discovery. We are researching ways of using IRs to develop domain-targeted drugs, especially for cancer treatment. In addition, we are developing a cancer model mouse having a target protein that can be controlled in a reversible fashion.
About Our Research
Puromycin technology developed in Japan
Around 1995, some researchers reported that ribonucleic acids (RNA) could acquire functions by directed evolution in a test tube ("in vitro evolution"). The next target of in vitro evolution was to make proteins evolve faster into preferred forms in the test tube. As a result, we succeeded in developing the in vitro virus (IVV) method (Figure 1A) as a research tool for analyzing the in vitro evolution of proteins. In addition, we developed a method of labeling the C-terminus of a protein using puromycin (Figure 1B). This method was fortuitously discovered while developing the IVV method. We named these two protein analysis methods "puromycin technology".
Figure 1. Puromycin Technology
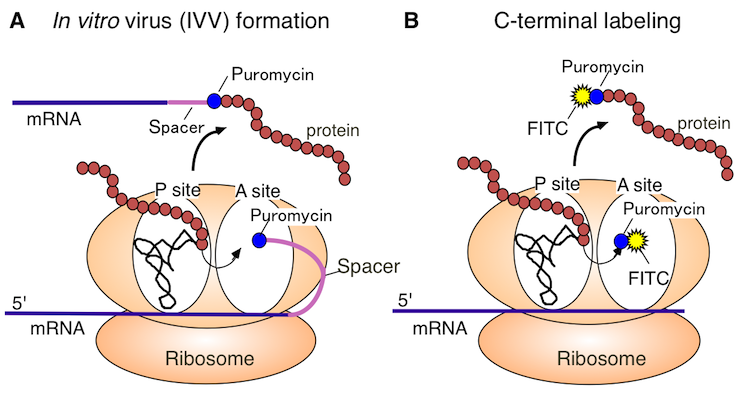
A. The figure is a schematic representation of the in vitro virus (IVV) formation. In vitro virus (IVV) is a fusion molecule of mRNA (genotype) and encoded protein (phenotype) covalently linked via puromycin and a polyethylene glycol spacer. The reaction of the IVV formation is catalyzed by the ribosome-centered translational machinery in a cell-free system.
B. The figure is a schematic representation of the protein C-terminal labeling procedure. Under moderately low puromycin concentration conditions, puromycin derivatives (e.g., fluorescent puromycin conjugate) bind to the C-terminus of the full-length protein. This reaction is catalyzed by the ribosome-centered translational machinery in a cell-free system.
Functional analysis in the post-genomic era: The world's first analysis tool for region-level protein interactions
In 2000, when the research on puromycin technology was still in its infancy (Miyamoto-Sato et al., NAR, 2000), a historical event in life science took place. In June of that year, Dr. Francis Collins, a director of international human genome project (HGP), formally declared that mapping of 90% of the regions of the human genome was completed. Scientists worldwide were excited by this announcement, which heralded the opening of the 'post-genomic' era for unraveling the code written in the genomic sequences. In response to this, the Japanese government started some national projects related to the human genome.
At the same time, we started applying the puromycin technology to the functional analysis of proteins in the post-genomic era. In studies dealing with a large amount of data like genomic sequences, not only experimental analysis tools, but also interdisciplinary approaches, including bioinformatics, were needed. In 2005, we succeeded in developing a high-throughput system for protein interaction analysis through collaborative research with the IT company (Miyamoto-Sato et al., Genome Res., 2005).
Yeast two-hybrid (Y2H) system and TAP (Tandem Affinity Purification)-MS (mass spectrometry) methods are well-known PPI analysis methods. However, these methods suffer from certain problems, such as cell toxicity and the library size derived from cell manipulations in their experimental procedures. Our methods can overcome these problems.
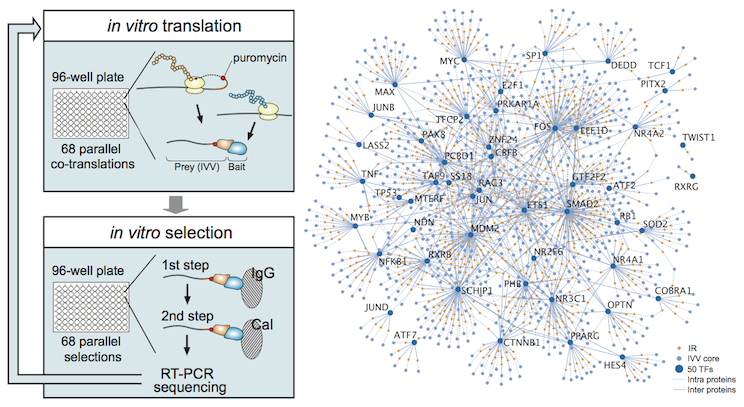
In the genome network project (promoted as a national project in Japan), we developed an automated system that could conduct large-scale experiments using the IVV method (Figure 2-left). Using this system, we succeeded in the world's first region-/domain-level interaction analysis of human transcription factors (Figure 2-right; Miyamoto-Sato et al., PLoS One, 2010). The data is freely available on the website (Genome Network Platform) of National Institute of Genetics.
Figure 2. The analysis of transcription factor networks using the IVV method